
In RNA-Seq analyses, adding pre-determined quantity of synthetic RNA sequences (spike-ins) to samples is a popular way to verify the experimental pipeline, determine quantification accuracy and for normalisation of differential expression. The most commonly used spike-ins are the ERCC spike-ins.
This post will cover the bioinformatic steps involved in obtaining read counts of spike-ins from a FASTQ file sequenced with spike-ins. The steps are namely creating a custom FASTA genome build incorporating the spike-in sequences, custom GTF file creation, mapping the reads to the custom genome, read counting and visualisation. This post will not be covering the wet lab part of adding spike-ins. I have a FASTQ data file (sample01.fq.gz) from single cell 50bp single-end Illumina reads with spike-ins that I am using for this workflow.
Go to the manufacturer’s website and obtain the Spike-in sequences. Mine was obtained from Thermofisher. Download the sequence file.
wget "https://tools.thermofisher.com/content/sfs/manuals/cms_095047.txt"
We can take a look at the file.
column -s, -t < cms_095047.txt | less -#2 -N -S
1 ERCC_ID GenBank 5prime_assay 3prime_assay Sequence
2 ERCC-00002 DQ459430 Ac03459872_a1 Ac03459968_a1 TCCAGATTACTTCCATTTCCGCCCAAGCTGCTCACAGTATACGGGCGTC
3 ERCC-00003 DQ516784 Ac03459873_a1 Ac03459969_a1 CAGCAGCGATTAAGGCAGAGGCGTTTGTATCTGCCATTATAAAGAAGTT
4 ERCC-00004 DQ516752 Ac03459874_a1 Ac03459970_a1 TCTTGCTTCAACAATAACGTCTCTTTCAGAAGGCATTGGTATCTTTTCC
5 ERCC-00009 DQ668364 Ac03459876_a1 Ac03459972_a1 CAATGATAGGCTAGTCTCGCGCAGTACATGGTAGTTCAGCCAATAGATG
6 ERCC-00012 DQ883670 Ac03459877_a1 Ac03459973_a1 CGAGAGATGTTTGTAGGTGCGGAATGTGTGCGGTCTACCTTAGCTGTAG
7 ERCC-00013 EF011062 Ac03459878_a1 Ac03459974_a1 ACCGAGCTCAGATGTGAAGGATCTTCTTGGAGGATTAAAAAAATGATTA
8 ERCC-00014 DQ875385 Ac03459879_a1 Ac03459975_a1 AAAAGAAGAAGGAAAGTAGAGGAGATCAAGATGTCAAACGAAACAATTA
9 ERCC-00016 DQ883664 Ac03459880_a1 Ac03459976_a1 GTTGTAACGAATGTTAATTTAGGAGGCAAGAGTTTGTGGGCGCGGACTT
10 ERCC-00017 DQ459420 Ac03459881_a1 Ac03459977_a1 GAGAACTGAAAGTGAGTCCCAACGAGAGAGGTGCATCTGTCCAGTGAGA
11 ERCC-00019 DQ883651 Ac03459883_a1 Ac03459979_a1 CTTTAATGGTTGTACATACTTGACGGATTGCAGTGAGTGTATTCCCGTC
12 ERCC-00022 DQ855004 Ac03459884_a1 Ac03459980_a1 CCCGGGCCAATTGCCTCTATAACTAGAGCTGAGCCCACCATTAAAGCGA
13 ERCC-00024 DQ854993 Ac03459886_a1 Ac03459982_a1 ACGGAGGAGCTTTGGCATACTAGGCTAGCGAATCTGCAACTAACGCAAG
14 ERCC-00025 DQ883689 Ac03459887_a1 Ac03459983_a1 GGGGTCCATTATAGTGCAGGCGTGGTAAAGTAGCATTAGAACCCTACTA
15 ERCC-00028 DQ459419 Ac03459888_a1 Ac03459984_a1 AAGAAAGTATTCCATTCCGGCTCATGGTCCCGGCTAGACCTGCAAGATC
16 ERCC-00031 DQ459431 Ac03459889_a1 Ac03459985_a1 GGGAGAAAAGCTTCTTGCCTAGTTACAGCACGAAGATTGGGACCCATCG
It is a tabular format with two columns that we need: ERCC ID and Sequence.
Genome file
The first step is to convert this tabular file into FASTA format and then append that to our genome FASTA file. We convert this table into a FASTA format file named ercc.fa.
cat cms_095047.txt | awk -F "\t" -v OFS="" 'NR>1{print ">",$1," ",$2," ",$3," ",$4,"\n",$5}' > ercc.fa
I am working with (Zebrafish) Danio rerio GRCz10. Let’s take a look at the Zebrafish FASTA genome file.
tail -n 20 Danio_rerio.GRCz10.dna.toplevel.fa
>CACACACACACACACACACACACACACACACACACATACACACTCACACAACGCAAACGG
ACATACAGTACACAAACACACTCAAGACACCACAGACCAAACACACACACACTCCAAACA
CAATAGACAATATTGACTATAATAGCCAATAATCTGTCCCACAATCCCAAACACCGACAG
CACTGCTGGATTACAGCCAATAATGCAACAACTCTGGAAGGAAAAGCCTGCAGGTCAGAG
GCCTTTAATAATGTAGATCACACTTAACCCGGTTAGAGAGGCTAATGGGGTCAAACAGGC
CTGTGTGGCATTTTACATCACCAGGAAACCACTGGAGAACATTTAACACACACACACACA
CACACACACACACAAGCAGATGTGCACATATACACATACATACATTATATATGTATACAT
ACATACAT
>KN150525.1 dna:scaffold scaffold:GRCz10:KN150525.1:1:650:1 REF
GTGTATTATAAAACATACGAGACTTAAAGTATAATCTATCCAGCACCTATAGAGCCACTC
GCGTTGCTTACGAAATTAGTATTTAGAAAGACCCACCACAAAGAAAAGAGATGTGTCTGT
ATGTATAAAAAATATATGAGACAGGGTTATAACCACCCGACGCGCTTTGTCTAAATAGAA
GTCGAGCACACACACACACACACACACACACACACACACACACACACACGGGTCAGCCAG
AGGACAGCTAACCTGCACACACCAGTCAAACCATGAACGGCAGAGAAACACCTCCATAAT
CCACACTCACCCCTGACGAGCACACACACACACACACACATATACACACACACACACACA
CTATAGTAAACTGGGTGTGCGTGGATGTGTGTATGTGTGTGATGGACTAGGATGTGTGTG
TGTGTGTGTGTGTGTGTGTGTGTGTGTGTGTGAGCTCACGTTCACTGAAGACGAAGTCTG
CGTTGATGTCGAAGGGCTGGATCTGGATGTCGTAGATCGACACGGTGACGCCTTTCTGAT
GGTCGCTGGAGATTAGAGTCAGCGTCTGCTGCGCCGCACACACATAGGAGCGCCCGGCCG
GAGTCACCAGCGCCGAGAGACGGTGTGTGCTGGCAGTGTGTTTCCCCGCT
We make a copy of the genome.
cp Danio_rerio.GRCz10.dna.toplevel.fa Danio_rerio.GRCz10.dna.toplevel.ercc.fa
And add the contents of ercc.fa file to the bottom of the copied Zebrafish genome file.
cat ercc.fa >> Danio_rerio.GRCz10.dna.toplevel.ercc.fa
Now, our new genome FASTA file looks like so
tail -n 5 Danio_rerio.GRCz10.dna.toplevel.ercc.fa
>ERCC-00170 DQ516773 Ac03459966_a1 Ac03460062_a1
TATTGGTGGAGGGGCACAAGTTGCTGAAGTTGCGAGAGGGGCGATAAGTGAGGCAGACAG
GCATAATATAAGAGGGGAGAGAATTAGCGTAGATACTCTTCCAATAGTTGGTGAAGAAAA
TTTATATGAGGCTGTTAAAGCTGTAGCAACTCTTCCACGAGTAGGAATTTTAGTTTTAGC
TGGCTCTTTAATGGGAGGGAAGATAACTGAAGCAGTTAAAGAATTAAAGGAAAAGACTGG
CATTCCCGTGATAAGCTTAAAGATGTTTGGCTCTGTTCCTAAGGTTGCTGATTTGGTTGT
TGGAGACCCATTGCAGGCAGGGGTTTTAGCTGTTATGGCTATTGCTGAAACAGCAAAATT
TGATATAAATAAGGTTAAAGGTAGGGTGCTATAAAGATAATTTAATAATTTTTGATGAAA
CCGAAGCGTTAGCTTTGGGTTATGAAACTCCATGATTTTCATTTAATTTTTTCCTATTAA
TTTTCTCCTAAAAAGTTTCTTTAACATAAATAAGGTTAAAGGGAGAGCTCTATGATTGTC
TTCAAAAATACAAAGATTATTGATGTATATACTGGAGAGGTTGTTAAAGGAAATGTTGCA
GTTGAGAGGGATAAAATATCCTTTGTGGATTTAAATGATGAAATTGATAAGATAATTGAA
AAAATAAAGGAGGATGTTAAAGTTATTGACTTAAAAGGAAAATATTTATCTCCAACATTT
ATAGATGGGCATATACATATAGAATCTTCCCATCTCATCCCATCAGAGTTTGAGAAATTT
GTATTAAAAAGCGGAGTTAGCAAAGTAGTTATAGACCCGCATGAAATAGCAAATATTGCT
GGAAAAGAAGGAATTTTGTTTATGTTGAATGATGCCAAAATTTTAGATGTCTATGTTATG
CTTCCTTCCTGTGTTCCAGCTACAAACTTAGAAACAAGTGGAGCTGAGATTACAGCAGAG
AATATTGAAGAACTCATTCTTTAGATAATGTCTTAGGTTAAAAAAAAAAAAAAAAAAAAA
AAA
>ERCC-00171 DQ854994 Ac03459967_a1 Ac03460063_a1
CTGGAGATTGTCTCGTACGGTTAAGAGCCTCCGCCCGTCTCTGGGACTATGGACGGGCAC
GCTCATATCAGGCTATATTTGGTCCGGGTTATTATCGTCGCGGTTACCGTAATACTTCAG
ATCAGTTAAGTAGGGCCATATGCCTCGGGAATAAGCTGACGGTGACAAGGTTTCCCCCTA
ATCGAGACGCTGCAATAACACAGGGGCATACAGTAACCAGGCAAGAGTTCAATCGCTTAG
TTTCGTGGCGGGATTTGAGGAAAACTGCGACTGTTCTTTAACCAAACATCCGTGCGATTC
GTGCCACTCGTAGACGGCATCTCACAGTCACTGAAGGCTATTAAAGAGTTAGCACCCACC
ATTGGATGAAGCCCAGGATAAGTGACCCCCCCGGACCTTGGAGTTTCATGCTAATCAAAG
AAGAGCTAATCCGACGTAAAGTTGCGGCGTTGATTACGCAGGATTGCGACCAAAGAACGA
GAAAAAAAAAAAAAAAAAAAAAAAA
Annotation file
Now we define the annotation file. Our original Ensembl GTF file looks like this
tail -n 5 Danio_rerio.GRCz10.84.gtf
>KN150216.1 ensembl exon 353 443 . - . gene_id "ENSDARG00000099551"; gene_version "1"; transcript_id "ENSDART00000165750"; transcript_version "1"; exon_number "3"; gene_name "tgds"; gene_source "ensembl"; gene_biotype "protein_coding"; transcript_name "tgds-201"; transcript_source "ensembl"; transcript_biotype "protein_coding"; exon_id "ENSDARE00001154426"; exon_version "1";
KN150216.1 ensembl CDS 353 443 . - 0 gene_id "ENSDARG00000099551"; gene_version "1"; transcript_id "ENSDART00000165750"; transcript_version "1"; exon_number "3"; gene_name "tgds"; gene_source "ensembl"; gene_biotype "protein_coding"; transcript_name "tgds-201"; transcript_source "ensembl"; transcript_biotype "protein_coding"; protein_id "ENSDARP00000138327"; protein_version "1";
KN150216.1 ensembl exon 267 280 . - . gene_id "ENSDARG00000099551"; gene_version "1"; transcript_id "ENSDART00000165750"; transcript_version "1"; exon_number "4"; gene_name "tgds"; gene_source "ensembl"; gene_biotype "protein_coding"; transcript_name "tgds-201"; transcript_source "ensembl"; transcript_biotype "protein_coding"; exon_id "ENSDARE00001169286"; exon_version "1";
KN150216.1 ensembl CDS 267 280 . - 2 gene_id "ENSDARG00000099551"; gene_version "1"; transcript_id "ENSDART00000165750"; transcript_version "1"; exon_number "4"; gene_name "tgds"; gene_source "ensembl"; gene_biotype "protein_coding"; transcript_name "tgds-201"; transcript_source "ensembl"; transcript_biotype "protein_coding"; protein_id "ENSDARP00000138327"; protein_version "1";
KN150216.1 ensembl five_prime_utr 938 942 . - . gene_id "ENSDARG00000099551"; gene_version "1"; transcript_id "ENSDART00000165750"; transcript_version "1"; gene_name "tgds"; gene_source "ensembl"; gene_biotype "protein_coding"; transcript_name "tgds-201"; transcript_source "ensembl"; transcript_biotype "protein_coding";
We will make a copy of the GTF file.
cp Danio_rerio.GRCz10.84.gtf Danio_rerio.GRCz10.84.ercc.gtf
We will create similar GTF file from the ERCC tabular file. Each ERCC sequence is defined as a new chromosome. First we define them as genes with gene_id staring with G followed by the ERCC ID and biotype ‘ercc’. This is written to a file ercc.gtf
cat cms_095047.txt | awk -F "\t" -v OFS="" 'NR>1{print $1,"\t","ercc","\t","gene","\t","1","\t",length($5),"\t",".","\t","+","\t",".","\t","gene_id \"G",$1,"\"; ","gene_version \"1\"; ", "gene_name \"",$1,"\"; ","gene_source \"ercc\"; ","gene_biotype \"ercc\";"}' > ercc.gtf
The ercc.gtf now looks like this:
head ercc.gtf
>ERCC-00002 ercc gene 1 1062 . + . gene_id "ERCC-00002"; gene_version "1"; gene_name "ERCC-00002"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00003 ercc gene 1 1024 . + . gene_id "ERCC-00003"; gene_version "1"; gene_name "ERCC-00003"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00004 ercc gene 1 524 . + . gene_id "ERCC-00004"; gene_version "1"; gene_name "ERCC-00004"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00009 ercc gene 1 985 . + . gene_id "ERCC-00009"; gene_version "1"; gene_name "ERCC-00009"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00012 ercc gene 1 995 . + . gene_id "ERCC-00012"; gene_version "1"; gene_name "ERCC-00012"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00013 ercc gene 1 809 . + . gene_id "ERCC-00013"; gene_version "1"; gene_name "ERCC-00013"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00014 ercc gene 1 1958 . + . gene_id "ERCC-00014"; gene_version "1"; gene_name "ERCC-00014"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00016 ercc gene 1 845 . + . gene_id "ERCC-00016"; gene_version "1"; gene_name "ERCC-00016"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00017 ercc gene 1 1137 . + . gene_id "ERCC-00017"; gene_version "1"; gene_name "ERCC-00017"; gene_source "ercc"; gene_biotype "ercc";
ERCC-00019 ercc gene 1 645 . + . gene_id "ERCC-00019"; gene_version "1"; gene_name "ERCC-00019"; gene_source "ercc"; gene_biotype "ercc";
The ercc.gtf is appended to the bottom of the copied Zebrafish GTF file.
cat ercc.gtf >> Danio_rerio.GRCz10.84.ercc.gtf
Mapping
We create a genome index as normal. I am using STAR aligner.
star \
--runMode genomeGenerate \
--runThreadN 8 \
--genomeFastaFiles /home/ngs/raw/Danio_rerio.GRCz10.dna.toplevel.ercc.fa \
--genomeDir /home/ngs/index/starindex-ercc/
And then we map our single-end or paired-end FASTQ files using the new ERCC genome index and ERCC GTF file. Here I have a single-end read gzipped FASTQ file.
star \
--genomeDir "/home/ngs/index/starindex-ercc/" \
--runThreadN 12 \
--readFilesCommand zcat \
--readFilesIn "sample01.fq.gz" \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix "sample01-" \
--sjdbGTFfile "/home/ngs/raw/Danio_rerio.GRCz10.84.ercc.gtf"
This should produce a BAM file that can be used in the next step.
Quantification
The reads can be quantified (read counts) using any of the standard workflows. Reads can be quantified from the BAM files using HTSeq count, featureCounts etc. or the FASTQ files can be used directly with KALLISTO. Here I use QoRTs. QoRTs is a versatile tool for gene and transcript quantification as well as RNA-Seq QC. Chromosome-level counts, gene-level counts, transcript counts, biotype counts and numerous other results can be obtained.
QoRTs can be run as below for single-end reads. Note that --maxReadLength must be specified for single-end reads. I had to specify --maxPhredScore because my phred scores exceed the value of 41. --minMAPQ is used to specify that only uniquely mapped reads must be counted. Max mapping quality (unique mapping) for STAR is the value 200.
java -Xmx20G -jar /home/programs/qorts/qorts-1.1.2.jar QC \
--singleEnded \
--generatePlots \
--generateMultiPlot \
--noGzipOutput \
--verbose \
--maxReadLength 43 \
--minMAPQ 60 \
--maxPhredScore 100 \
"sample01.bam" \
"/home/raw/Danio_rerio.GRCz10.84.ercc.gtf" \
"sample01-qorts"
I have also run featureCounts for comparison.
subread-1.5.0-p2-Linux-x86_64/bin/featureCounts \
-a "/home/raw/Danio_rerio.GRCz10.84.ercc.gtf" \
-o "sample01-featurecounts-gene.txt" \
-F "GTF" \
-t "exon" \
-g "gene_id" \
-M \
-p \
-C \
-B \
-S rf \
-T 6 \
"sample01.bam"
Here are some of gene counts (QC.geneCounts.txt) for Spike-ins from the QoRTs results. The second column contains the counts we are looking for.
GENEID COUNT COUNT_CDS COUNT_UTR COUNT_AMBIG_GENE
GERCC-00002 2062 0 2062 0
GERCC-00003 24 0 24 0
GERCC-00004 411 0 411 0
GERCC-00009 0 0 0 0
GERCC-00012 0 0 0 0
GERCC-00013 0 0 0 0
GERCC-00014 0 0 0 0
GERCC-00016 0 0 0 0
And here are some of the gene counts from featureCounts. The last column contains the counts we need.
Geneid Chr Start End Strand Length sample01.bam
GERCC-00002 ERCC-00002 1 1062 + 1062 2062
GERCC-00003 ERCC-00003 1 1024 + 1024 24
GERCC-00004 ERCC-00004 1 524 + 524 413
GERCC-00009 ERCC-00009 1 985 + 985 0
GERCC-00012 ERCC-00012 1 995 + 995 0
GERCC-00013 ERCC-00013 1 809 + 809 0
GERCC-00014 ERCC-00014 1 1958 + 1958 0
GERCC-00016 ERCC-00016 1 845 + 845 0
Visualisation
We can confirm that everything has worked and the ERCC sequences are mapping to the custom genome build by inspecting the resulting BAM file. The BAM file can be visualised in a genome browser such as IGV. I am using SeqMonk. I created a new genome in SeqMonk using the customised ERCC FASTA genome file and the custom ERCC GTF file. I then imported my BAM file. Below is a preview from SeqMonk.
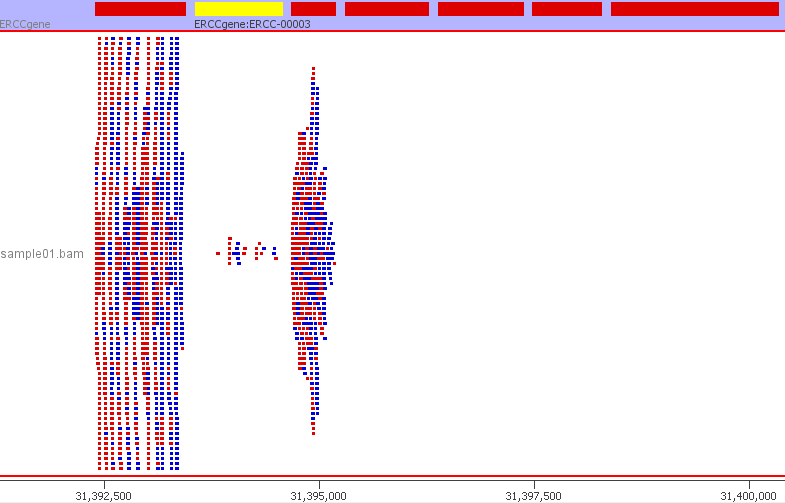

The SeqMonk visualisation adds up well with the read counts from QoRTs and featureCounts. For example, the 24 reads mapping to ERCC-00003. The read counts are now ready to be used in downstream analyses such as differential gene expression using DESeq2, EdgeR or Limma. The ERCC ID counts will be considered just like any other gene.



Comments